GOLDENRING LABORATORY RESEARCH
The origin of pre-neoplastic metaplasia in the stomach
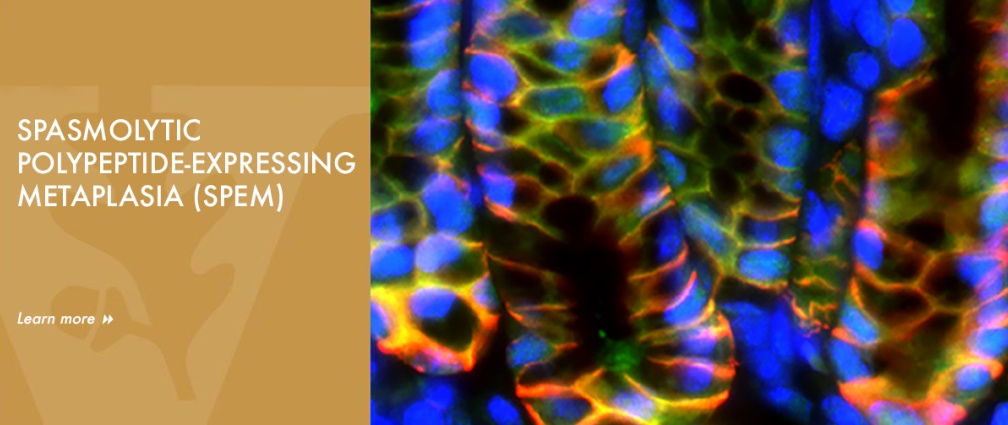
The Goldenring lab studies of pre-neoplastic changes related to gastric cancer and the roles of mucosal stem cells in giving rise to metaplasia and cancer. Dr. Goldenring first described Spasmolytic Polypeptide-expressing Metaplasia (SPEM) in both mouse models and humans in 1999. While those initial studies were controversial, work over the past 5 years has now established the validity of the concept that SPEM is present in many rodent models of parietal cell loss and in humans. The Goldenring lab has redefined concepts of the origin of metaplasia and altered views of the plasticity of differentiated cells by demonstrating, using lineage mapping techniques, that SPEM arises from transdifferentiation of mature chief cells into mucous cell metaplasia. All of these studies have led to a major change in the paradigm for gastric preneoplasia to recognize a pathway from transdifferentiation of chief cells into SPEM in the presence of parietal cell loss to further progression of metaplasia into a proliferative metaplasia under the influence of inflammatory mediators. Transdifferentiation of chief cells into SPEM is an orderly process, which requires down-regulation of the zymogenic machinery in chief cells, upregulation of mechanisms to deal with oxidative stress, autophagy of zymogen granules, and reprograming of the transcriptome to adopt a mucous cell phenotype. The Goldenring lab defined M2-macrophages, IL-13 and IL-33 as critical inflammatory mediators of induction of metaplasia and progression of metaplasia towards more proliferative and intestinalized pre-neoplastic lineages. Ongoing studies now seek to evaluate the role of ILC2 cells as obligate intramucosal immune cells regulating induction of metaplasia. In the past 4 years, The Goldenring lab has provided evidence that activation of Ras in chief cells (using the Mist1-Kras(G12D) mouse) can lead to all of the metaplastic changes observed in humans, including development of SPEM, followed by evolution of intestinal metaplasia and further development of dysplasia. Just as exciting they have shown that treatment of either the Mist1-Kras(G12D) mice or Mongolian gerbils infected with H. pylori for 12 months with a MEK inhibitor causes arrest of metaplasia and reestablishment of normal gastric lineages. These findings have major implications for possible reversal of metaplasia in patients at high risk for gastric cancer development.
Apical membrane recycling, epithelial polarity, congenital diarrheal diseases
Dr. Goldenring has been a leader in the investigation of the roles of specific roles of Rab small GTPases in regulating vesicle trafficking and membrane recycling in polarized cells. Dr. Goldenring first discovered Rab11a on parietal cell proton pump-containing tubulovesicle recycling membranes. He utilized yeast two-hybrid screening to identify multiple Rab11a interacting proteins including Myosin Vb (MYO5B) and the Rab11-Family Interacting Proteins (Rab11-FIPs). Over the past 2 decades, the Goldenring lab has worked in a number of epithelial cell systems to investigate the role of Rab11a and its interacting proteins in epithelial cell physiology. These studies established that phosphorylation of Rab11-FIP1 and Rab11-FIP2 by the polarity associated kinase MARK2/Par1b, which regulates both epithelial cell polarity and junction integrity.
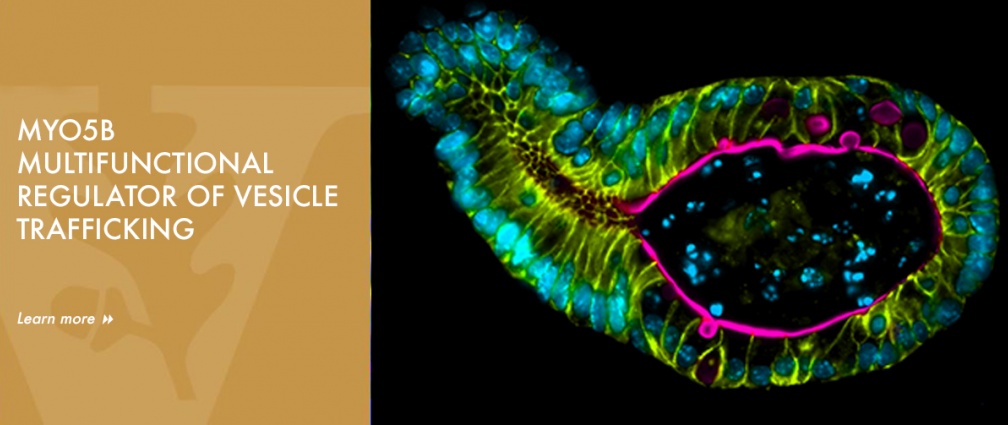
The Goldenring lab established MYO5B as a multifunctional regulator of vesicle trafficking. They demonstrated that MYO5B can bind all of the Rab11 family members (Rab11a, Rab11b and Rab25) as well Rab8a and Rab10. More recently, the lab has focused on how alterations in regulators of apical membrane recycling system can lead to changes in the integrity of the intestinal enterocyte brush border. Their most recent studies have focused on the pathophysiology of inactivating mutations in MYO5B in Microvillus Inclusion Disease (MVID) and the characterization of novel mouse models of MYO5B deletion. These studies have led to the recognition that MYO5B regulates multiple trafficking pathways to the apical membrane that must be traversed by transporters on their way to the brush border. In particular, loss of MYO5B in MVID leads to deficits in apical NHE3 and SGLT1, which are required for apical fluid absorption. They also have revealed that loss of MYO5B leads to formation of microvillus inclusions through an unrecognized apical bulk endocytosis mechanism that is dependent on dynamin 2 and pacsin 2. He has also broadened his studies to consider the identification of genetic congenital causes of undiagnosed neonatal diarrheas and approaches to treatment of these maladies. These studies have recently led to the first detailed evaluation of apical trafficking defects in children with mutations in DGAT1. All of these cell biology and cell physiology studies seek to define common and distinct vesicle trafficking pathways among different epithelial cells.
Ongoing studies in mouse models as well as mouse and pig enteroid systems, seek to elucidate the molecular mechanisms of apical bulk endocytosis and the complexity of vesicle trafficking and recycling systems involved in apical membrane transport. Additionally, ongoing investigations seek to identify therapeutic options for bypassing the blockade in apical trafficking in MYO5B deficient enterocytes and in other congenital enterocyte abnormalities.