Skip to main content
Giving
Careers
Patient Care
Referring Providers
Price Transparency
Employee Resources
MyWorkday
search
Search
What are you looking for?
Search
Search
search
Menu
open menu
Close
close menu
Search
Patient Care
Patient Care Home
Urologic Oncology
Endourology and Stone Disease
Pediatric Urology
Reconstructive Urology and Pelvic Health
General Urology
Our People
Education
Education Home
Residency
Residency Home
Our Residents
Endourology & Laparoscopic/Minimally Invasive Surgery Fellowship
Female Pelvic Medicine and Reconstructive Surgery Fellowship
Genitourinary Reconstruction and Trauma Fellowship
Pediatric Urology Fellowship
Urologic Oncology Fellowship
Urology Nurse Practitioner Fellowship
Medical Students
Medical Students Home
Visiting Rotations
Research/Innovation
Research/Innovation Home
Translational Research
Health Services
Clinical Trials
Pediatric Urology
Reconstructive Urology and Pelvic Health
Robotic Surgery, Medical Devices, and Image-Guided Surgery
Urologic Oncology
Urology Frontiers
Basic Science
About Us
About Us Home
History of Innovation
News and Events
Alumni
Contact Us
Giving
Giving Home
Give Now
Intranet
Refer a Patient
Vanderbilt University Medical Center
Monroe Carell Jr. Children's Hospital at Vanderbilt
Vanderbilt University
Research
For Patients and Visitors
Resources for Employees and Researchers
Giving
Careers
Patient Care
Referring Providers
Price Transparency
Employee Resources
MyWorkday
Department of Urology
Patient Care
Urologic Oncology
Endourology and Stone Disease
Pediatric Urology
Reconstructive Urology and Pelvic Health
General Urology
Our People
Education
Residency
Our Residents
Endourology & Laparoscopic/Minimally Invasive Surgery Fellowship
Female Pelvic Medicine and Reconstructive Surgery Fellowship
Genitourinary Reconstruction and Trauma Fellowship
Pediatric Urology Fellowship
Urologic Oncology Fellowship
Urology Nurse Practitioner Fellowship
Medical Students
Visiting Rotations
Research/Innovation
Translational Research
Health Services
Clinical Trials
Pediatric Urology
Reconstructive Urology and Pelvic Health
Robotic Surgery, Medical Devices, and Image-Guided Surgery
Urologic Oncology
Urology Frontiers
Basic Science
About Us
History of Innovation
News and Events
Alumni
Contact Us
Giving
Give Now
Intranet
Refer a Patient
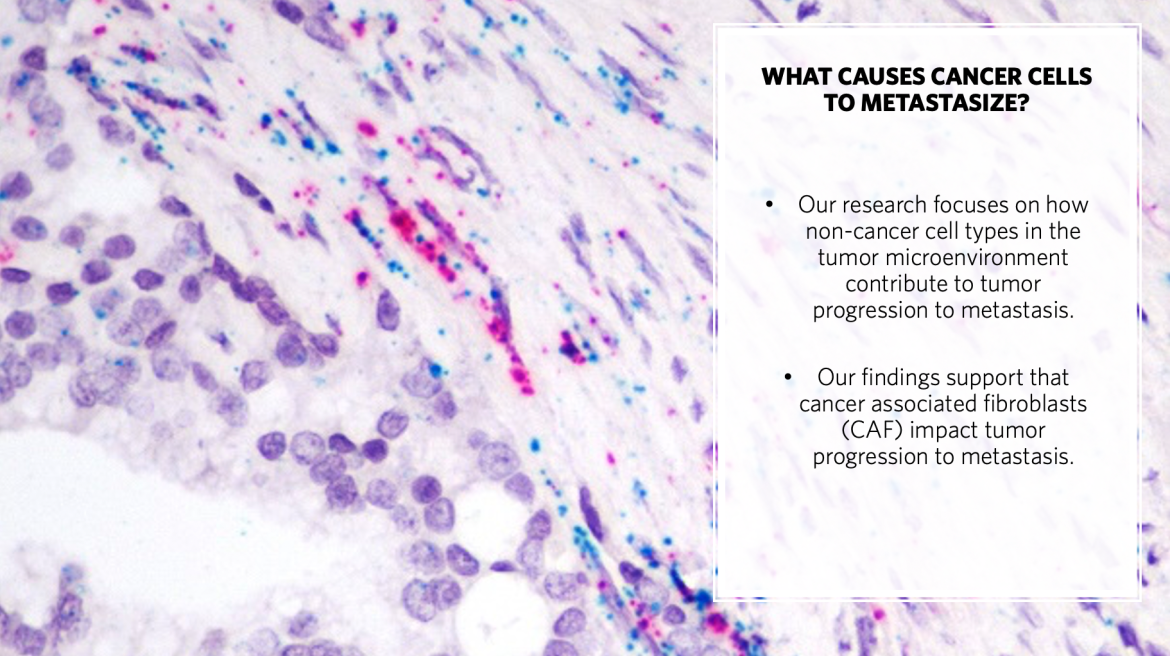
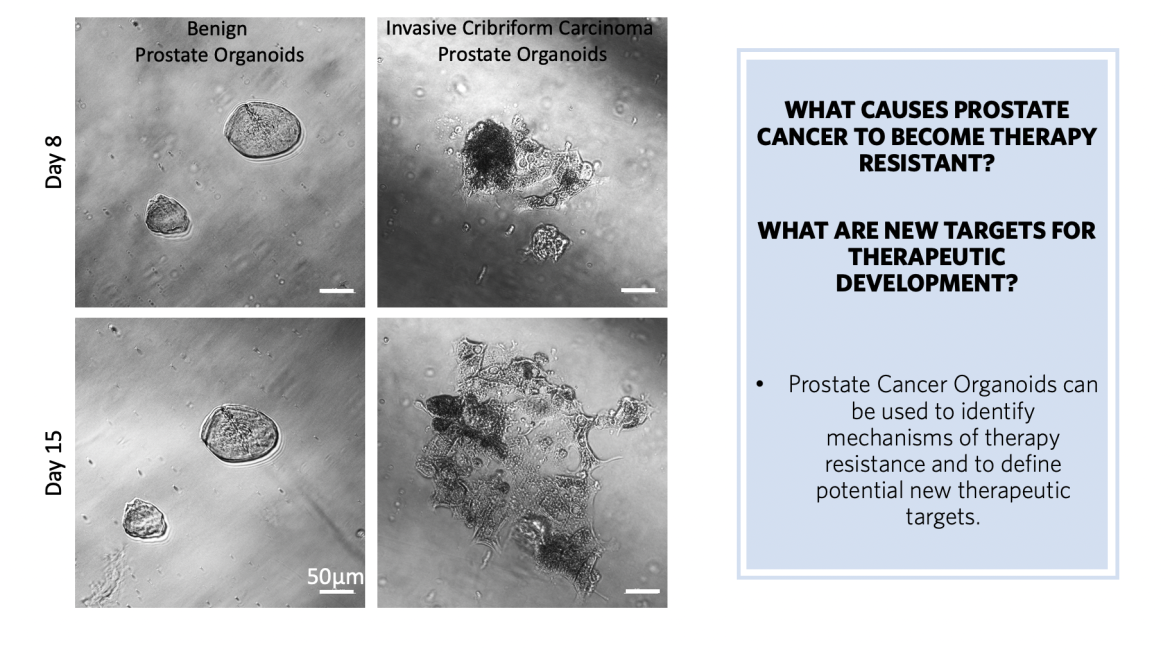
Innovation: Basic Science
Basic Science